FERI - Zvijanje proteinov
Zvijanje proteinov
Zvijanje proteinov je eden od največjih izzivov stoletja. Zaradi tega privablja raziskovalce iz različnih področij. Na področju računalništva raziskovalci izdelujejo algoritme, ki simulirajo zvijanje proteinov s pomočjo računalnika in tako napovedujejo nativno stanje proteina. Proteini so sestavljeni iz aminokislin in nativno stanje predstavlja tisto obliko oz. postavitev aminokislin v prostoru, ko protein izvaja svojo biološko funkcijo oz. ima najmanjšo energijo. V nasprotnem primeru lahko povzroča različne bolezni.
V okviru te naloge izdelajte program, ki bo uporabil poenostavljen model HP in poskušal poiskati čim boljšo postavitev aminokislin v 3D prostoru za podano zaporedje aminokislin. Zaporedje aminokislin je sestavljena iz črk. V primeru modela HP so aminokisline razdeljene v dve skupini H (hidrofobne) in P (hidrofilen). Podano zaporedje predstavlja povezano verigo aminokislin, ki jo je potrebno postaviti v kubično mrežo na tak način, da je njena energija čim manjša. Pri tem je potrebno upoštevati, da lahko eno aminokislino postavimo le v eno vozlišče kubične mreže. Energija pa predstavlja število hidrofobnih povezav med dvema aminokislinama, ki nista sosednji. Da dobimo končno vrednost energije, dobljeno število hidrofobnih povezav pomnožimo z -1. Npr. zaporedje HPHHPHPHH lahko postavimo v kubično mrežo tako, da zasede naslednja vozlišča: {(0, 0, 0), (0, 0, −1), (1, 0, −1),(1, −1, −1), (1, −1, 0), (1, 0, 0), (2, 0, 0), (2, 0, −1), (2, −1, −1)} in je prikazana na sliki. Vidimo, da so hidrofobne aminokisline predstavljene s temnejšimi in hidrofilne s svetlejšimi kroglicami. Na osnovi teh informacij lahko ugotovimo, da imamo 4 hidrofobnih povezav, kar pomeni, da je energija te postavitve enaka -4.
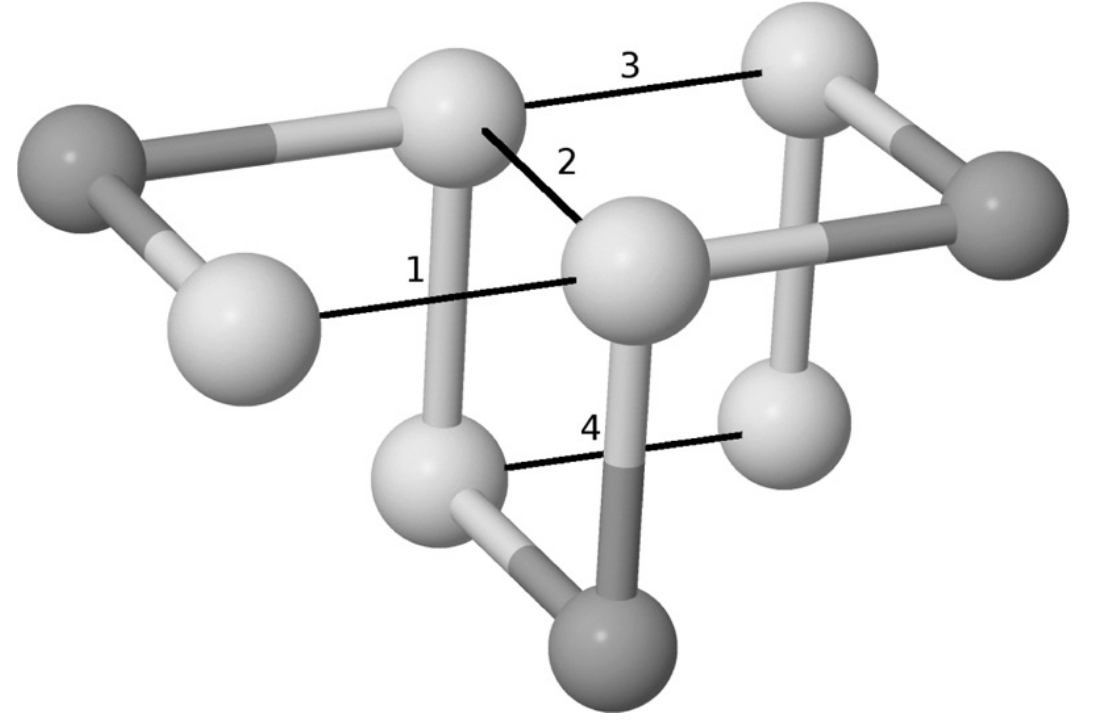
Torej cilj naloge je izdelati program, ki prejme zaporedje aminokislin modela HP in poskuša poiskati najboljšo možno njihovo postavitev znotraj kubične mreže. Boljša postavitev, je tista, ki ima več hidrofobnih povezav oz. manjšo energijo.
Strokovni mentor, kontakt: borko.boskovic@